Abstract
Thrombolysis is the current standard of care for ischemic stroke. In order to better understand its underlying mechanisms, a closer look at the basic biochemical processes involved in tissue repair and coagulation is necessary. Upon vascular injury, multiple stepwise pathways become activated in order to stop the bleeding and start the process of healing. Vessel wall recovery and coagulation (among other anatomical abnormalities) have been implicated to be the cause of stroke. Here, a basic biochemistry of hemostasis, blood coagulation, and endogenous anticoagulation mechanisms as well as pharmacokinetics of exogenous anticoagulation prime the reader for better understanding the idea behind thrombolytic therapy for stroke.
Contents:
1- Hemostasis and The Coagulation Cascade
1A. aPTT
1B. PT & INR
1C. TCT
2- Endogenous vs. Exogenous Anticoagulation and Antifibrinolytic Systems, and The Role of Tissue Plasminogen Activator (tPA)
1- Hemostasis and The Coagulation Cascade
Upon vascular injury, the first stage of healing and damage control is hemostasis. The interaction between platelets, coagulation and fibrinolytic factors as well as anti-inflammatory mediators and leukocytes complete the primary and secondary hemostasis4. In primary hemostasis, at the site of injury the smooth muscle in the vessel wall contracts (vasoconstriction). Platelets are then activated by chemicals released at the injury site (e.g., erythrocytes and white blood cells) and by contact with underlying collagen. The platelets then stick to one another at the wound site (platelet plug)6. In the meantime, in the secondary hemostasis, fibrinogen is converted to fibrin, forming a mesh that traps more platelets and erythrocytes, forming a clot (figure 1). At this stage, the anti-thrombotic control mechanism is also activated in order to counteract the adverse effects and control the clot formation and coagulation4,6.
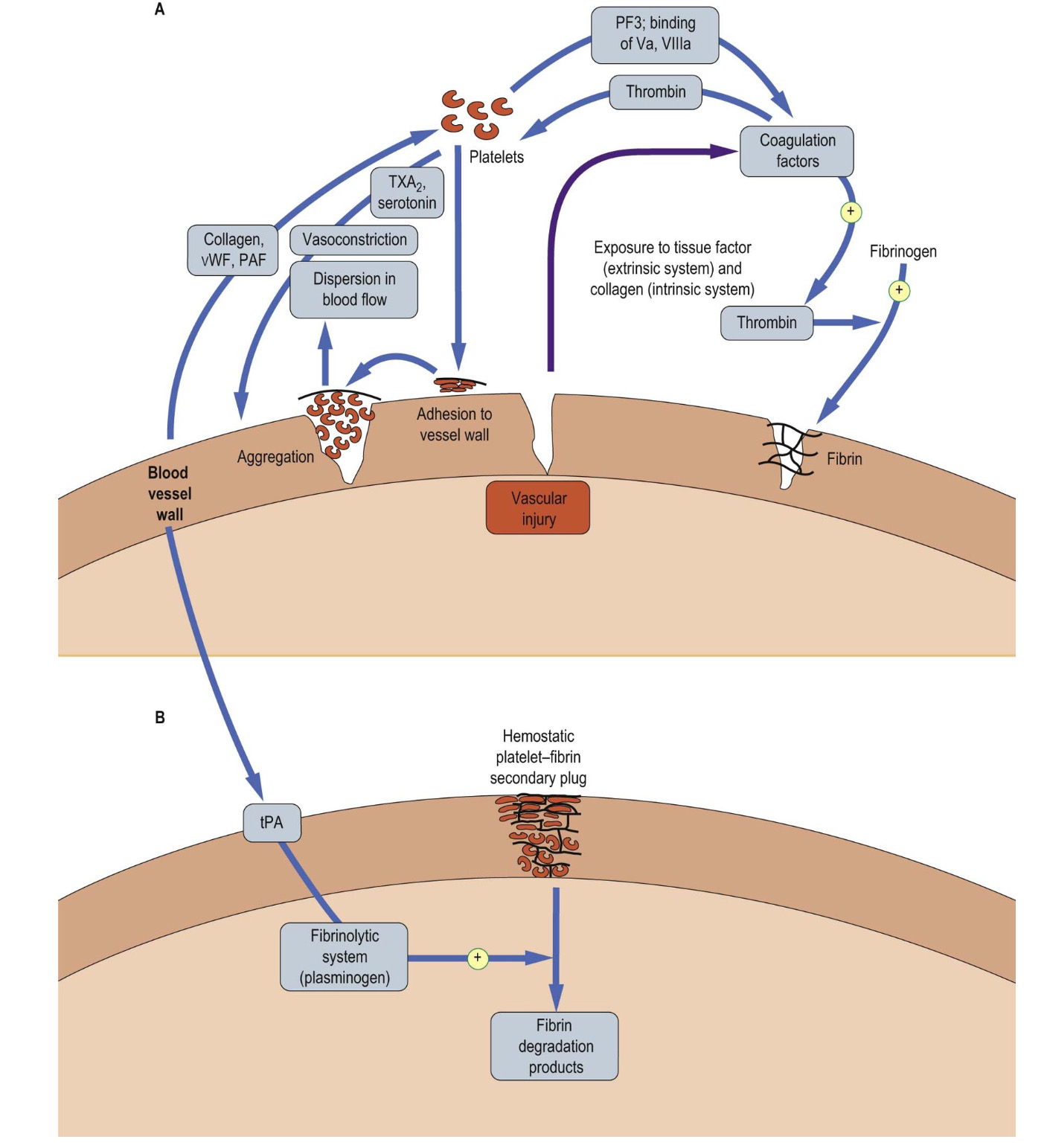
Figure 1: Hemostasis: A) vascular injury triggers a cascade of events which leads to a primary plug by platelets. B) The platelet plug is stabilized by a network of fibrin. the secondary plug is stable and is only degraded when the fibrinolytic system is activated (picture credit: Dominiczak MH. Medical Biochemistry. London: Elsevier, 2012).
The coagulation cascade (figure 2) is a complex and nonlinear process that is divided to three parts: 1) the intrinsic pathway, 2) the extrinsic pathway, and 3) the final common pathway8.
The three components of the coagulation system are defined based on the nature of initiating factors and the corresponding clinical hemostasis laboratory tests on citrated platelet-poor plasma:
- Testing the intrinsic pathway: activated partial thromboplastin time (aPTT)
- Testing the extrinsic pathway: prothrombin time (PT)
- Testing the final common pathway: thrombin clotting time (TCT)
The platelet-poor plasma is used in these tests because the platelet count influence clotting time results. To obtain platelet-poor plasma, blood is collected in tubes containing citrate anticoagulant to dampen calcium ion reversibly, and the blood is centrifuged at 2000g for 15 minutes. The coagulation time tests are initiated by adding calcium and appropriate initiating agents. However, these tests have their limitations in describing the in vivo phenotype of a patient’s blood to coagulate effectively6.
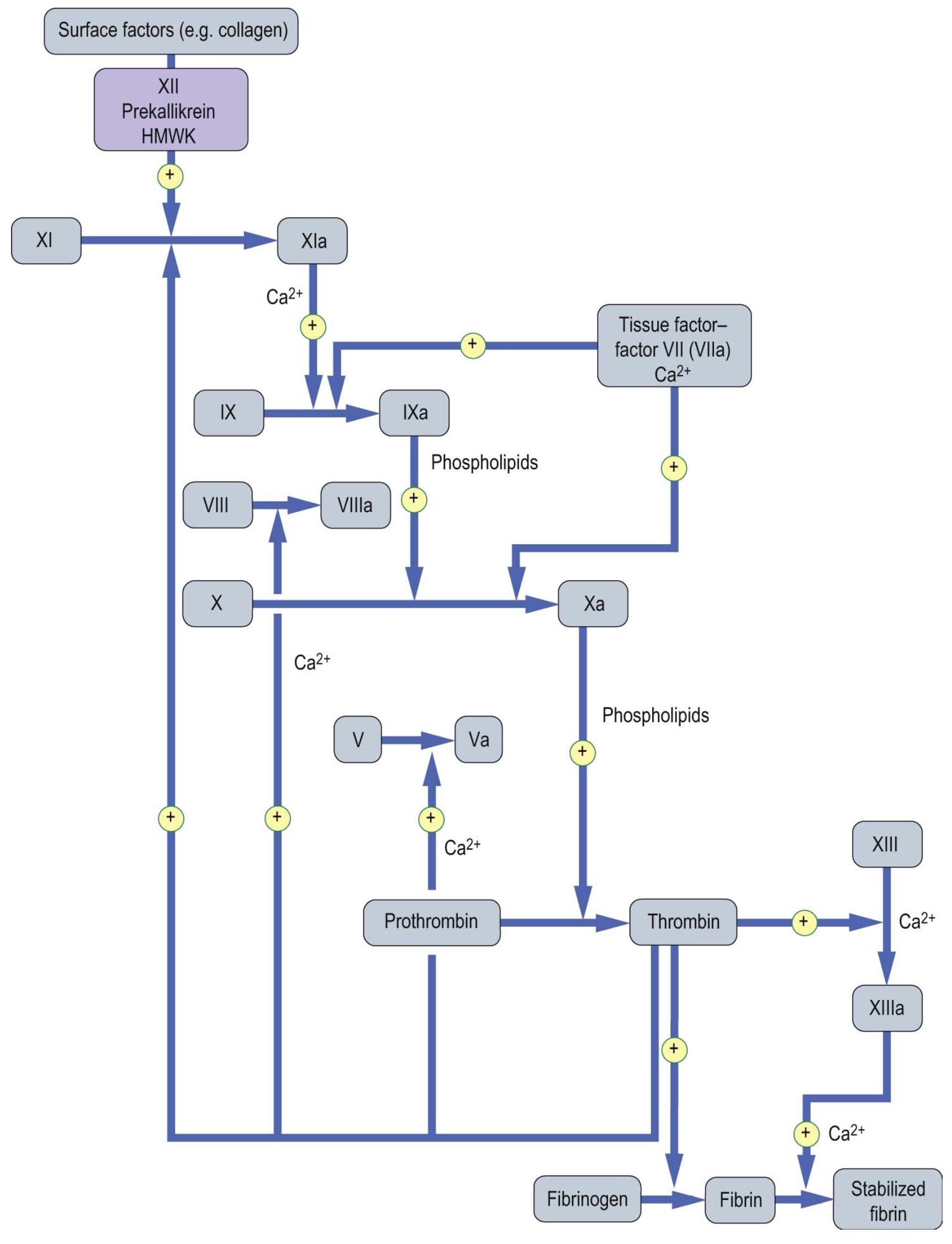
Figure 2: The coagulation cascade (picture credit: Dominiczak MH. Medical Biochemistry. London: Elsevier, 2012).
aPTT
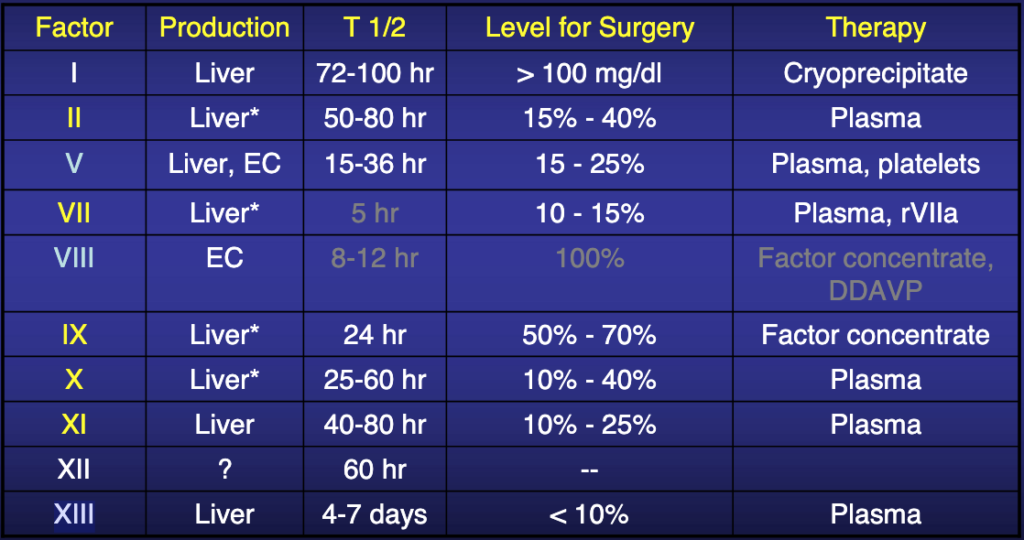
Activated partial thromboplastin time (aPTT)—also known as Kaolin-Cephalin clotting time (KCCT)— measures the intrinsic pathway. The term “intrinsic” signifies that no extrinsic factor such as tissue factor or thrombin is added to the blood. There reference range for the aPTT is about 30-40 seconds; prolongation observed in deficiencies of factors 12, 11, 9 or its cofactor, factor 8 (VIII) (consult table 1 for coagulation factors and their properties). This test is used to exclude the common congenital hemophilia—deficiencies of factors 8, 9, 11—and to monitor unfractionated heparin treatment4.
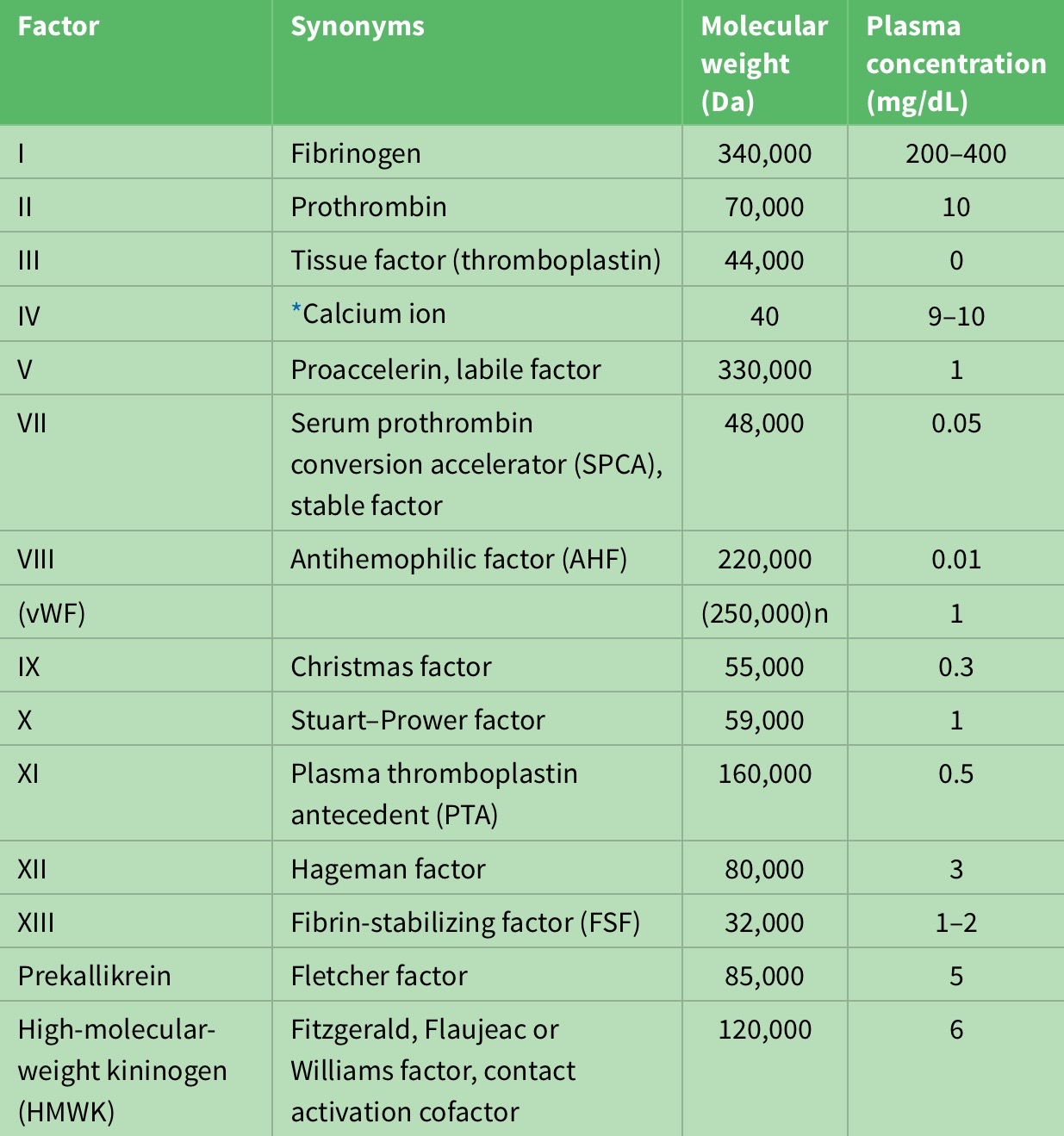
Table 1: Properties of coagulation factors.
PT & INR
Prothrombin time (PT) measures the extrinsic pathway, or the effect of tissue factor, which after combining with coagulation factor 7, greatly accelerates coagulation by activating both factor 9 and 104. Tissue factor is a polypeptide that is expressed in all cells other than endothelial cells. The reference range for PT is approximately 10-15 seconds; prolongation could be indicative of deficiencies in factors 2,5,7,10 in order to diagnose both the rare congenital defects of these factors and, much more commonly, acquired bleeding disorders, resulting from vitamin K deficiency (e.g, malabsorption or obstructive jaundice), administration of vitamin K antagonists ( e.g., warfarin) which reduce hepatic synthesis of these factors7.
Liver disease also reduces hepatic synthesis of the above factors. For example, the prothrombin time is a prognostic marker of liver failure after acetaminophen overdose (treatment is usually initiated by replacement therapy using fresh frozen plasma)4.
It is important to note that oral anticoagulants or vitamin K antagonists (warfarin) are usually prescribed for patients at risk of thrombosis—for example patients with atrial fibrillation or heart valve prostheses— and monitoring these patients every few weeks is essential to minimize the risk of thromboembolism and also of excessive bleeding8. The international normalized ration (INR) was developed and is calculated from a PT result. INR values between 2.0-3.0 (as shown in figure 3) are within the normal range, INR value above 3.0 indicates a higher risk of bleeding, and an INR value lower than 2.0 indicates a higher risk of clotting (e.g., stroke or venous thromboembolism)1,6.
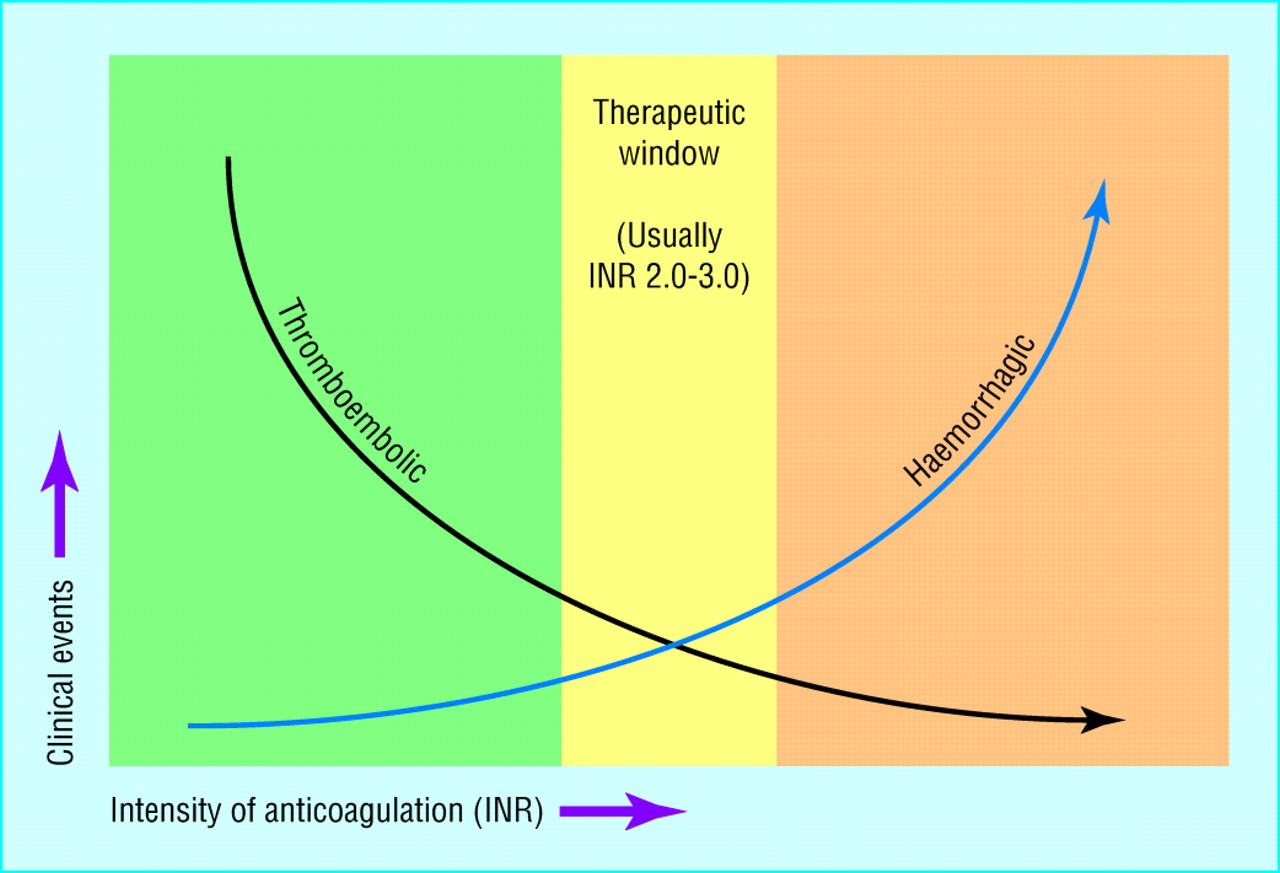
Figure 3 : Anticoagulation goal is confirmed by using INR values: the green area indicates a sub-therapeutic range which may lead to thromboembolic events, the yellow area is the optimal therapeutic goal and the orange signifies a high INR range which may lead to excessive bleeding1. (picture credit: Blann (2011))
Although many patients are still on warfarin and testing their coagulation levels is a necessary part of their therapeutic goal, in the past decade new direct oral anticoagulants such as dabigatran and rivaroxaban (mainly prescribed for atrial fibrillation) require no monitoring4.
TCT
Thrombin clotting time (TCT) asses the final common pathway which converts prothrombin to thrombin via factor 10a with factor 5a acting as a cofactor. This in turn yields the conversion of fibrinogen to fibrin. The reference range is approximately 10-15 seconds. TCT Prolongation is observed in fibrinogen deficiency and in the presence of inhibitors (e.g., heparin, fibrin degradation products). Fibrinogen deficiency may be congenital or due to acquired consumption of fibrinogen in disseminated intravascular coagulation (DIC) or may occur after administration of fibrinolytic drugs such as tissue plasminogen activator (tPA), streptokinase (SK) or urokinase (UK). Treatment is with cryoprecipitate or fibrinogen concentrates4,5,6. (consult table 3 in appendix for congenital and acquired causes of excessive bleeding)
Platelets
Aside from assessing platelet number, size and morphology through the “complete blood count” analysis and blood film review, platelet function can also be assessed (e.g., platelet function analyzer, light transmission aggregometry)
Endogenous vs Exogenous Anticoagulation and Antifibrinolytic Systems and The Role of Tissue Plasminogen Activator (tPA)
Coagulation inhibitors such as antithrombin (antithrombin III), protein C and S, and the tissue factor pathway inhibitor (TFPI) exist within the human body and are reared to as endogenous anticoagulants, which are mainly synthesized in the liver or in endothelium2,4,6. They are essential to prevent excessive thrombin formation and thrombosis (figure 4).
As mentioned earlier thrombin has a central role in hemostasis. Thrombin converts circulating fibrinogen to fibrin and activates factor 13, which crosslinks the fibrin to form a clot8. Also, thrombin stimulates its own generation in a positive feedback cycle by:
- catalysis of factor 11
- activation of factors 8 and 9
- activation of platelets
Due to thrombin’s central role in hemostasis and thrombosis, a number of novel oral direct thrombin inhibitors (DTIs) have been developed and a number of large randomized controlled clinical trials have confirmed the efficacy of DTIs4,9,10. Although lack of reversal agents has been the main area of concern, many clinicians have replaced warfarin by these novel DTIs (e.g., dabigatran). Another oral DTI that seems to be an effective alternative to heparin is argtroban, which has proven to be valuable, especially in cases where heparin is contraindicated due to heparin-induced thrombocytopenia1.
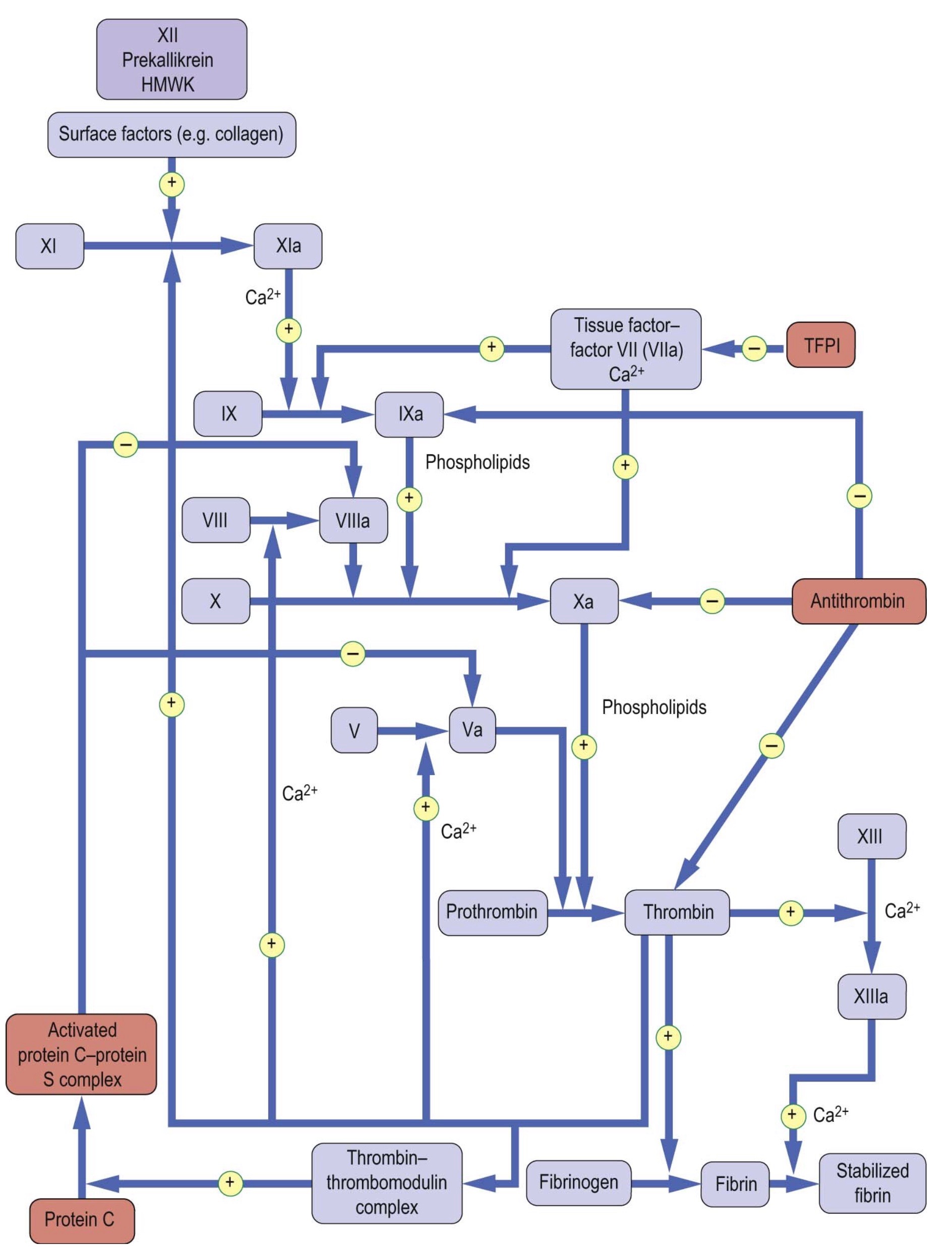
Figure 4: Anticoagulation factors and their sites of action (picture credit: Dominiczak MH. Medical Biochemistry. London: Elsevier, 2012).
On the other hand, the fibrinolytic system acts to limit excessive formation of fibrin through plasmin-mediated fibrinolysis. The coagulation system act s to form fibrin and the fibrinolytic system acts to limit excessive formation of fibrin both intra and extravascularly through plasmin-mediated fibrinolysis10. Circulating plasminogen binds to fibrin via lysine-binding sites; it is converted to active plasmin by plasminogen activators (see table 2). Tissue-type plasminogen activator (tPA) is synthesized by endothelial cells, but is released into plasma by stimuli such as venous occlusion, exercise and epinephrine2,4,6. Together with plasminogen, it binds strongly to fibrin, which stimulates its activity and localizing plasmin activity to fibrin deposits4.
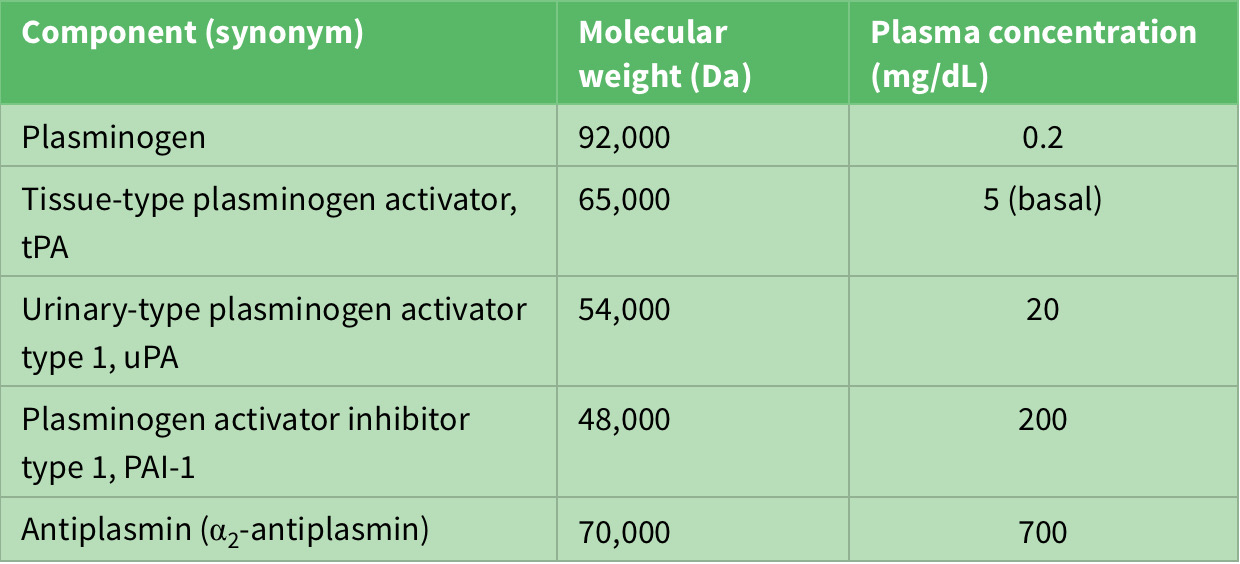
Table 2: Components of fibrinolytic system.
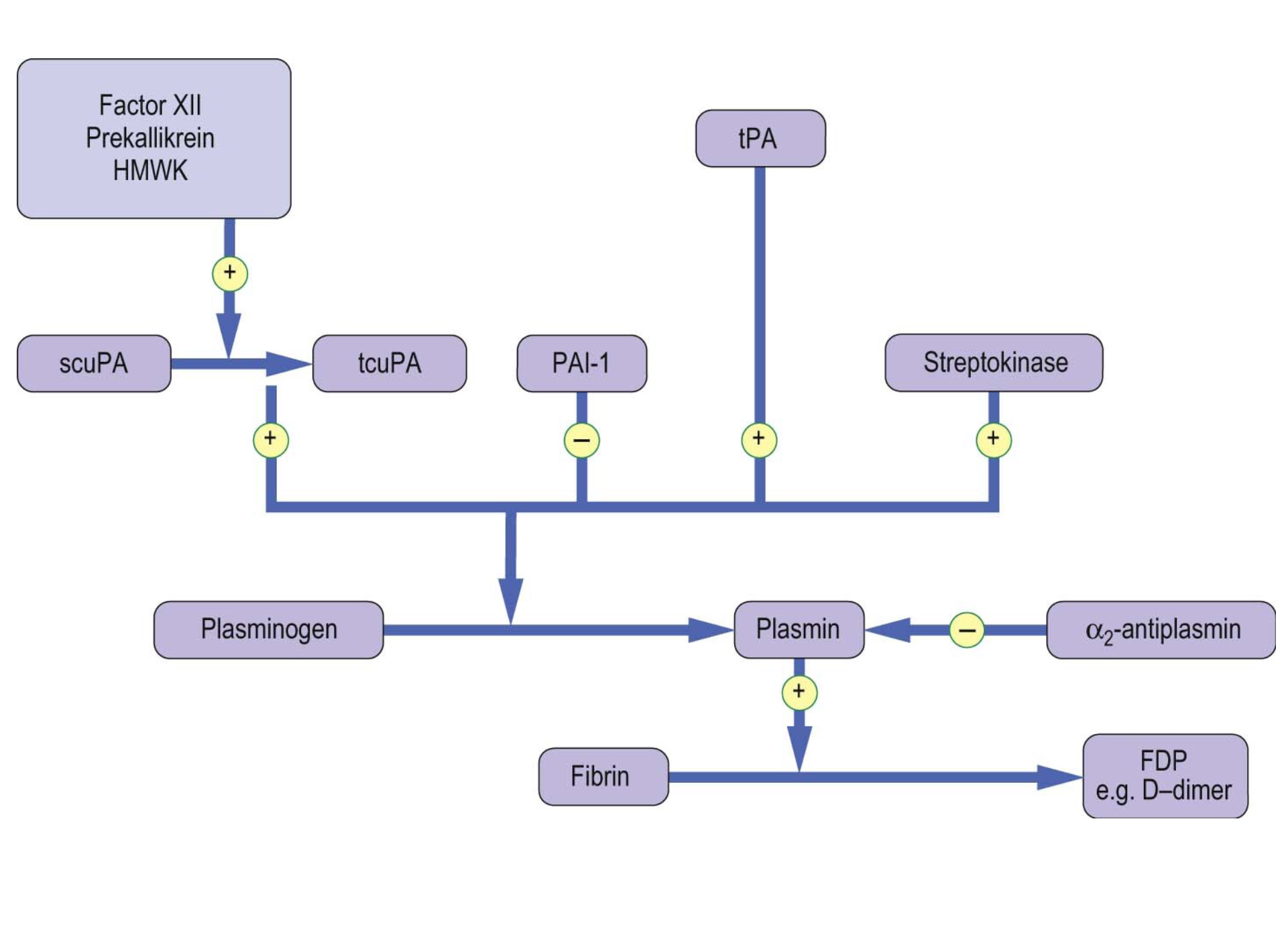
Figure 5: The fibrinolytic system. (picture credit: Dominiczak MH. Medical Biochemistry. London,: Elsevier, 2012).
Since the human body comprises agonist and antagonist system to maintain equilibrium, it is worth noting that plasmin inhibitors counteract excessive fibrinolytic activity. Plasminogen activator inhibitor type 1 (PAI-1) which is synthesized by both endothelial cells and hepatocytes inhibits excessive tPA activity. Urinary-type plasminogen activator (uPA) circulates in plasma both as an active single-chain precursor form (scuPA, pro-urokinase) and as more active two-chain form (tcuPA, urokinase). One activator of stupa is surface-activated coagulation factor 12, which therefore links the coagulation system to the fibrinolytic system6,9,10.
Despite controversies, in the past few decades pharmaceutical thrombolytics (fibrinolytics) such as urokinase and streptokinase used in reperfusion therapy, were tried with some degree of success in myocardial infarction, and in the past few years exogenous tPA agents such tenecteplase or alteplase have become the standard of care in both acute myocardial infarction and ischemic stroke2,3,7.In future posts the historical events leading to the discovery of the tPA, its application in management of stroke and current controversies will be explored.
Appendix:
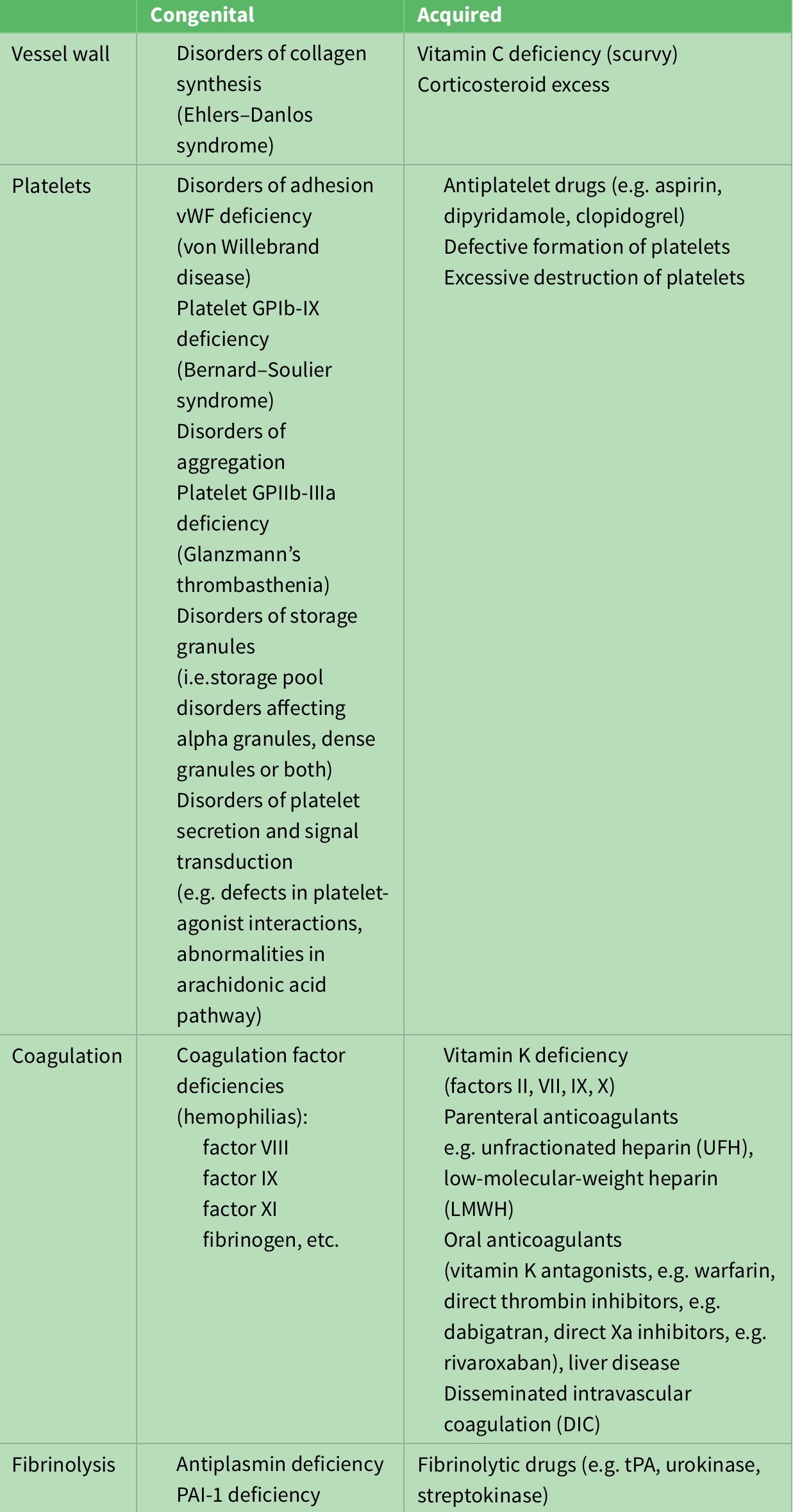
Table 3: Congenital and acquired causes of excessive bleeding.
References:
- Direct Thrombin InhibitorsBlann AD, Fitzmaurice DA, Lip GYH. Anticoagulation in hospitals and general practice. BMJ. 2003;326(7381):153-156. doi:1136/bmj.326.7381.153
- Campbell Bruce C.V., Meretoja Atte, Donnan Geoffrey A., Davis Stephen M. Twenty-Year History of the Evolution of Stroke Thrombolysis With Intravenous Alteplase to Reduce Long-Term Disability. Stroke. 2015;46(8):2341-2346. doi:10.1161/STROKEAHA.114.007564
- Campbell BCV, Mitchell PJ, Churilov L, et al. Tenecteplase versus Alteplase before Thrombectomy for Ischemic Stroke. New England Journal of Medicine. 2018;378(17):1573-1582. doi:10.1056/NEJMoa1716405
- Chaudhry R, Babiker HM. Physiology, Coagulation Pathways. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2019. http://www.ncbi.nlm.nih.gov/books/NBK482253/. Accessed July 5, 2019.
- Docagne Fabian, Parcq Jérôme, Lijnen Roger, Ali Carine, Vivien Denis. Understanding the Functions of Endogenous and Exogenous Tissue-Type Plasminogen Activator During Stroke. Stroke. 2015;46(1):314-320. doi:10.1161/STROKEAHA.114.006698
- Gale AJ. Current Understanding of Hemostasis. Toxicol Pathol. 2011;39(1):273-280. doi:10.1177/0192623310389474
- Parsons M, Spratt N, Bivard A, et al. A Randomized Trial of Tenecteplase versus Alteplase for Acute Ischemic Stroke. New England Journal of Medicine. 2012;366(12):1099-1107. doi:10.1056/NEJMoa1109842
- Smith SA, Travers RJ, Morrissey JH. How it all starts: initiation of the clotting cascade. Crit Rev Biochem Mol Biol. 2015;50(4):326-336. doi:10.3109/10409238.2015.1050550
- Weitz JI, Eikelboom JW, Samama MM. New Antithrombotic Drugs. Chest. 2012;141(2 Suppl):e120S-e151S. doi:10.1378/chest.11-2294
- Prevention and Treatment of Major Blood Loss | NEJM. https://www.nejm.org/doi/full/10.1056/NEJMra067742. Accessed July 5, 2019.